Regulatory Problems in Cancer Research
In this sequence I’ve been arguing that, while most cancer drugs developed over the past several decades are not very effective, there are potentially exciting avenues of research that haven’t gotten much attention or funding yet.
_Why _is this the case? Cancer research is a huge field full of intelligent people. Cancer is a very common disease and there’s a lot of money to be made in treating it. “Curing cancer” is a byword for a lofty goal. Why should there be any 20-dollar bills lying on the sidewalk at all?
In particular, why would there be less progress since the War on Cancer, which allocated much _more _federal funding to cancer research than was available before?
The conventional story is that cancer is simply hard. We already gathered the low-hanging fruit of radiotherapy and cytotoxic chemotherapy; now we’re trying to cure the tougher cancers, and it just takes more money and time.
I’ve been arguing that the “cancer is hard” story is incorrect. Targeted chemotherapy, the most popular approach for the past two decades, tends to fail because of the incredible diversity and mutability of cancer. Approaches that focus on what cancers have in _common, _like their high glucose requirements or susceptibility to immune defenses, might turn out to work much better.
But, if I’m correct, why hasn’t some enterprising cancer researcher already come to the same conclusions? Even if I’m wrong, I’m not unique; a lot of my argument is just echoing James Watson’s views. Why haven’t any investors and funders decided it might be a good idea to try cancer research the way the discoverer of the double helix thinks we should do it?
This can be explained by an increase in the regulatory burden of cancer research in the past several decades. Clinical trials have become more expensive, require more paperwork, and allow less freedom of judgment from clinicians and researchers. Only a large pharmaceutical company can afford to run Phase II and III clinical trials these days. There’s more money in cancer research than ever, but it’s harder to try new things on sick people. This tends to _narrow _cancer research to established players and drug classes.
In the world of early-stage tech startups, success follows a power-law distribution. Investors gain more money by funding a handful of huge successes than they lose by giving small investments to a lot of things that don’t work out. So it makes sense that, for instance, YCombinator keeps casting a wider net, accepting startups at earlier stages, and actively seeking outliers and mavericks. They want to make sure they don’t miss the next AirBnB.
It would seem to make sense that investing in drug candidates would work similarly; you don’t want to miss the next Gleevec either. But if the cost of testing is too high, casting a wide net becomes much more expensive. You can’t just give the founders of an early-stage biotech company a little funding to see if they can do something awesome with it. And so, medical research becomes much more conservative.
Regulation Has Increased Costs and Slowed Drug Development
90% or more of a typical drug’s costs come from Phase III clinical trials. So it makes sense to focus on the costs and barriers associated with clinical trials, to see if they’ve gone up over time and what the consequences have been.
As of 2005, the R&D cost of the average drug was $1.3 billion. In 1975, that figure was $100 million. (That is, drug trials have gotten on average more than 13 times more expensive over the past forty years.) Phase III trials are becoming longer, involving more procedures and more hours of work, and have lower enrollment and retention due to more stringent enrollment criteria and trial protocols.
Protocols for clinical trials are now over 200 pages long on average. The combined costs are $26,000 per patient. The annual rate of cost increase is itself increasing, from an annual increase of 7.3% in 1970-1980 to 12.2% from 1980-1990 (inflation-adjusted.) The estimated cost per life-year saved from current clinical cancer trials is approximately $2.7 million.
It now takes an average of 12-15 years from drug discovery to marketing, compared to an average of 8 years in the 1960’s. Before the 1962 Kefauver-Harris amendment that vastly increased FDA powers, it took only 7 months. For oncology drugs, just the preclinical work takes 6 years; once early clinical trial data are in, it takes 26-27 months to proceed to Phase II or III; and it takes an average of 14.7 clinical trials (Phase I, II, or III) to get a drug approved.
Running a clinical trial requires protocols to be approved by the FDA, the NCI (National Cancer Institute, the primary funder of cancer research in the US), and various IRBs (institutional review boards, administered by the OHRP, or Office of Human Research Protections.) On average, “16.8% of the total costs of an observational protocol are devoted to IRB interactions, with exchanges of more than 15,000 pages of material, but with minimal or no impact on human subject protection or on study procedures.” Adverse events during trials require a time-consuming reporting and re-consent process. While protocols used to be guidelines for investigators to follow, they are now considered legally binding documents; so that, for instance, if a patient changes the dates of chemotherapy to schedule around family or work responsibilities, that is considered to be a violation of protocol that can void the whole trial.
To handle this regulatory burden, an entire industry of CROs (contract research organizations) has grown up, administering trials and handling paperwork to make the experimental drug look good to federal regulators. Like tax preparers, CROs have an incentive to keep the regulatory process complex and expensive.
The result of all this added cost is that fewer drugs get developed than otherwise would. Sam Peltzman’s 1973 study of drug availability and safety before and after the 1962 Kefauver-Harris amendment (which significantly enhanced FDA powers) found that a model of drug development predicted a post-1962 average of 41 new drugs approved per year, while the actual average was 16 new drugs approved per year. The pre-1962 average number of drugs approved per year was 40.
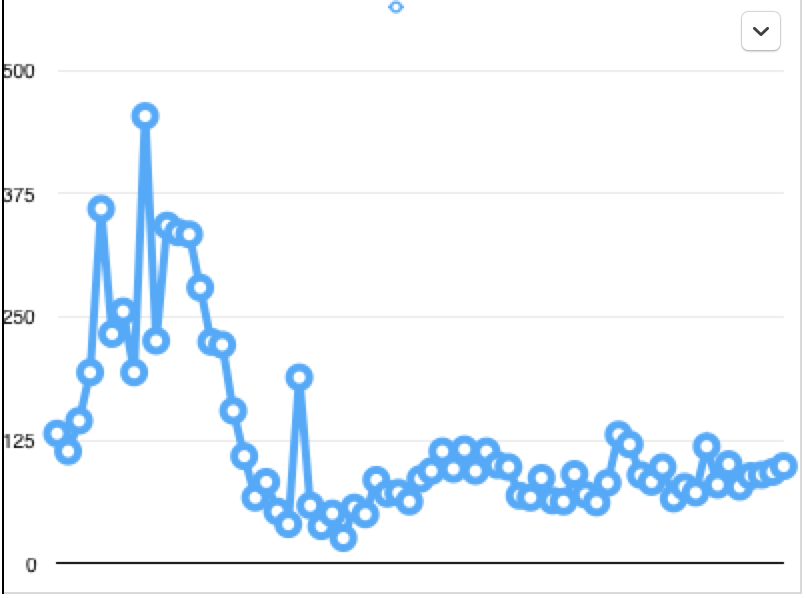
This is a graph of the number of new drug applications approved by the FDA every year from 1944 to the present. Note that the number of drugs approved has been largely flat since the 1962 Kefauver-Harris amendment, though the decline in drug approvals appears to precede the law by several years.
Increased Drug Regulation Has Not Meaningfully Decreased Risk
Peltzman’s study on the Kefauver-Harris amendment found that there was little evidence suggesting that more ineffective drugs reached the market pre-1962 compared to post-1962.
Comparing the US to Great Britain and Spain, each of which approve more drugs per year than the US, the other countries have no higher rates of postmarket withdrawals of drugs, suggesting that the extra regulatory scrutiny is not providing us with safer drugs.
Toxic death rates haven’t dropped much in Phase I trials. In 6639 patients, comprising 211 trials, between 1972 and 1987, the toxic death rate was 0.5%. In 11,935 patients, comprising 460 studies, between 1991 and 2002, the toxic death rate was also 0.5%.
Between 1999 and 2006, the number of adverse drug reactions recorded in the US has actually been increasing, particularly as the proportion of elderly patients taking many drugs has increased.
The most common severe drug interactions are often from old, well-known drugs, like insulin, warfarin, and digoxin. “Antibiotics, anticoagulants, digoxin, diuretics, hypoglycaemic agents, antineoplastic agents and nonsteroidal anti-inflammatory drugs (NSAIDs) are responsible for 60% of ADRs leading to hospital admission and 70% of ADRs occurring in hospital.” Increasing regulation on new drugs isn’t going to stop the problem of increasing adverse drug reactions, because most of those come from old drugs.
Cost-Benefit Tradeoffs Support Looser Regulations On Drugs
Gieringer’s 1985 study estimated the loss of life from FDA-related delay of drugs since 1962 to be in the hundreds of thousands. This only includes the delay of drugs that were eventually approved, not the potentially beneficial drugs that were never approved or never developed, so it’s probably a vast underestimate.
In a recent paper, “Is the FDA Too Conservative Or Too Aggressive?“, the authors apply a Bayesian decision analysis to evaluate the overall cost of a trial based on the disease burden of Type I vs. Type II errors.
The classical approach used by the FDA is to constrain experiments to a maximum 2.5% risk of Type I error for all tests, and then choose a power for the alternative hypothesis by making the sample size large enough. That is, no drug can be approved if there is a greater than 2.5% chance that it is ineffective.
This doesn’t make sense from a disease risk standpoint, because for very severe diseases, the risk of not trying a drug that might work is higher than the risk of trying a drug that doesn’t work. The authors use data from the U.S. Burden of Disease study, which measures Years Lived with Disability to compute the “optimal” level of acceptable risk of inefficacy for drugs for different diseases. For instance, in pancreatic cancer, the BDA-optimal risk of Type 1 error is 27.9%, since the disease is so deadly.
Cancer in general, being both common and deadly, is an especially good area for looser drug regulation. If a new therapy increased the cure rate of lung cancer by just 1% (through improved adjuvant therapy) and increased the average life expectancy of uncured patients by just 3 months, the [five-year] regulation-induced delay would cost more than 2,000,000 life-years worldwide.
Even this cost-benefit framing may be understating the case for FDA and OHRP reform, though. The problem seems to be less that the standards for efficacy are too high, than that the costs of compliance are too high because of redundant and excessive required documentation. It would in principle be possible to streamline the process of conducting clinical trials without reducing its rigor.
We Think About Risk Wrong
In medical contexts, people often talk about the unknown as disrespectable. An “unapproved” drug, an “untested” drug, an “unproven” drug, a treatment that is “not indicated”, all sound unsettling. Nobody wants to play cowboy in life-and-death situations.
But this kind of language is not actually about reducing risk. _Reality is probabilistic; all choices have potential risks and potential benefits. There’s no _real wall, out in the universe, between the “safe/known” and the “unsafe/unknown”; that’s a human framing, akin to the Ellsberg paradox or the bias of ambiguity aversion. People prefer known risks to unknown risks.
In other words: death and disease are scary, and rightly so, but people will tend to be less frightened of risks that seem normal and natural (people have always died of cancer) than of risks that seem outlandish or like somebody’s fault (taking an experimental drug that might or might not work). Chosen risk, conscious risk, stepping into the unknown, is viewed as worse than the risk of passively allowing harm to occur. Even if the objective risk-benefit calculations actually work out the other way.
This is an instinct worth fighting. Cancer is a common disease, yes; but the “normalcy” of it can blind us to the horrifying death toll. As Bertrand Russell said, the mark of a civilized man is the capacity to read a column of numbers and weep.
Fear of action isn’t actually about making people safer. It’s about making people feel safer, because they aren’t looking at the whole picture. It’s about making people feel like they can’t be blamed.
It’s Too Hard to Do Transformative Biomedical Research Today
Derek Lowe, an always insightful observer of the pharmaceutical scene, comments on the VC firm Andreessen Horowitz’s first foray into biotech, “In this business, you work for years before you can have the tiniest hope of ever selling anything to anyone. And before you can do that, you have to (by Silicon Valley standards) abjectly crawl before the regulatory agencies in the US and every other part of the world you want to sell in. Even to get the chance to abase yourself in this fashion, you have to generate a mountain of carefully gathered and curated data, in which every part of every step must be done just so or the whole thing’s invalid, go back and start again and do it right this time. The legal and regulatory pressure is, by Valley standards, otherworldly.”
It shouldn’t be.
I am not a policy expert, so I don’t know what the appropriate next steps are. What kinds of reforms in FDA and OHRP rules have a reasonable chance of being passed? I don’t know at this point, and I hope some of my readers do.
I do know that committed activists can change things. In 1992, after a decade of heroic advocacy by AIDS patients, the FDA created the “accelerated approval” process, which can approve drugs for life-threatening diseases after Phase II studies.
We have to find a way to continue that legacy.